Research and Articles


Hotline
- Capital Markets Hotline
- Companies Act Series
- Climate Change Related Legal Issues
- Competition Law Hotline
- Corpsec Hotline
- Court Corner
- Cross Examination
- Deal Destination
- Debt Funding in India Series
- Dispute Resolution Hotline
- Education Sector Hotline
- FEMA Hotline
- Financial Service Update
- Food & Beverages Hotline
- Funds Hotline
- Gaming Law Wrap
- GIFT City Express
- Green Hotline
- HR Law Hotline
- iCe Hotline
- Insolvency and Bankruptcy Hotline
- International Trade Hotlines
- Investment Funds: Monthly Digest
- IP Hotline
- IP Lab
- Legal Update
- Lit Corner
- M&A Disputes Series
- M&A Hotline
- M&A Interactive
- Media Hotline
- New Publication
- Other Hotline
- Pharma & Healthcare Update
- Press Release
- Private Client Wrap
- Private Debt Hotline
- Private Equity Corner
- Real Estate Update
- Realty Check
- Regulatory Digest
- Regulatory Hotline
- Renewable Corner
- SEZ Hotline
- Social Sector Hotline
- Tax Hotline
- Technology & Tax Series
- Technology Law Analysis
- Telecom Hotline
- The Startups Series
- White Collar and Investigations Practice
- Yes, Governance Matters.
- Japan Desk ジャパンデスク
Pharma & Healthcare Update
February 17, 2020Dawn of a New Era: All Medical Devices in India to be Regulated from April 2020
- The Ministry of Health and Family Welfare has released two notifications to regulate all medical devices in India under the Medical Device Rules, 2017 from April 01, 2020.
- By way of the notifications, a catch-all definition will be notified under the Drugs and Cosmetics Act, 1940 which will cover all medical devices, thereby requiring the manufacturers, importers and sellers of the medical devices to obtain permission to engage in the import, manufacture and sale of the medical devices.
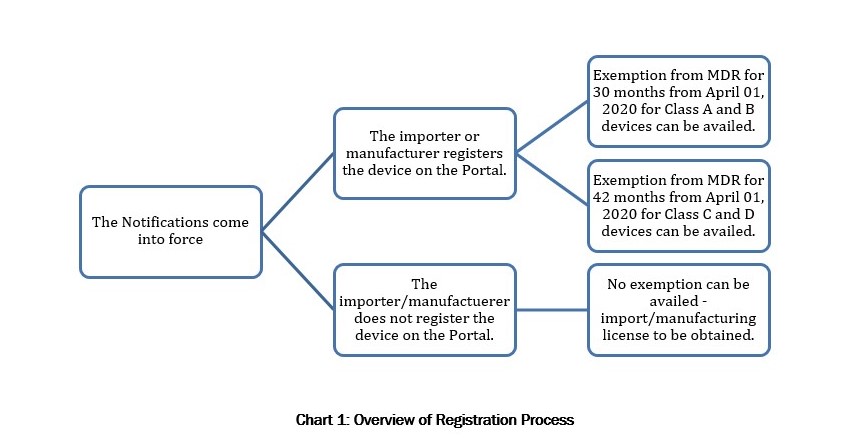
- Further, all devices (except those already regulated) will be required to register themselves online on a portal developed by the drug regulator for this purpose. The registration will be on a voluntary basis for the first 18 months and compulsory thereafter.
- Once a medical device has been registered on the portal, the medical device will be exempt from all regulatory compliances under the Medical Device Rules, 2017 for a period of 30 months from the date of the notifications for Class A and Class B devices and a period of 42 months from the date of the notifications in case of Class C and Class D devices.
- However, if a newly notified medical device is not registered, the provisions of the Medical Device Rules will apply with immediate effect.
A. INTRODUCTION
The Ministry of Health and Family Welfare (“Health Ministry”) on February 11, 2020 published two notifications, the first effectively notifying all medical devices by way of an expansive and catch-all definition of medical devices (“New Definition Notification”)1, and the second requiring the registration of such newly notified medical devices on a portal (“Portal”) developed by the Central Drugs Standard Control Authority (“CDSCO”) – India’s apex drug regulator (“Registration Notification”)2 (collectively referred to as the “Notifications”). The Notifications will come into effect on April 01, 2020.
The Health Ministry had released a draft version of the Notifications for public comments on October 18, 2019 (“Draft Notifications”). However, there are no changes between the Draft Notifications and the Notifications.
In this hotline, we have summarized the key features of the Notification. We have also analyzed the impact of the Notifications on the industry and our expectations on how the Notification should be enforced.
B. BACKGROUND
1.Medical Device Regulation
Medical devices in India are governed under the Medical Device Rules, 2017 (“MDR”) – a set of rules framed under India’s primary drug control legislation, the Drugs and Cosmetics Act, 1940 (“D&C Act”) – which came into force on January 01, 2018. The MDR only regulates medical devices which have been notified by the Health Ministry for regulation under the MDR. Currently, sixteen medical devices are regulated under MDR while 8 others are regulated as drugs. Four additional medical devices will come under regulation from January 01, 2021, eight more from April 01, 2021 and ultrasound equipment will be added to the list from November 01, 2020. The complete list of medical devices is provided below in Annexure A.
The MDR provides for a risk-based classification system with four categories from A to D for low risk, low moderate risk, moderate high risk and high risk respectively.3 Devices in classes C and D are regulated more stringently that devices in classes A and B as the former are considered to be high risk devices.
The slow pace and method of medical device regulation has been a concern for the industry for a while now. There are over 1700 types of medical devices in the global market4, out of which only 29 would have been governed under MDR by April 2021 if the Notifications had not been issued. It is also not feasible for the Health Ministry to notify all 1700 medical devices to bring them under the MDR. To remedy this, the Health Ministry has released the Notifications to regulate all medical devices in a phase-wise manner within 3.5 years from when the Notifications comes into force.
2.Impact of a Medical Device being Regulated under the MDR
The implications of being regulated under the MDR (“Regulated Device”) are manifold. We have provided an overview of the major implications of becoming a Regulated Device below.
- Additional Compliances under the MDR
The MDR divides the regulation of medical devices into the following broad activities – clinical investigation, clinical performance evaluation, import, manufacture and sale. At each stage of the production to consumption cycle, appropriate permissions are required to be obtained from the CDSCO (or other relevant state authority, as the case may be) in respect of the relevant activity. To elaborate, different permissions (in the form of licenses) are granted for importing medical devices, for conducting clinical investigations/clinical performance evaluations, and for manufacture and sale of such devices. Therefore, the level of compliance required to be undertaken by a manufacturer or importer of a Regulated Device will increase significantly. A detailed analysis of the requirements of the MDR are covered in our paper on the medical device industry, available here .
- Quality Control Requirements
In addition to procuring the appropriate licenses, the MDR also imposes strict quality control requirements on medical devices. Regulated Devices are required to follow standards set by the Bureau of India Standards (“BIS”). In the absence of BIS standards, manufacturers of Regulated Devices should follow standards laid down by the Health Ministry, the International Standards Organization or the International Electrotechnical Commission in that order.5 The MDR also prescribes a Quality Management System to be followed by manufacturers of Regulated Devices.6
- Price Control
All drugs (which include notified medical devices) are subject to price control in some form under the Drugs (Prices Control) Order, 2013 (“DPCO”). Therefore, Regulated Devices would also be subject to price control under the DPCO, either by being restricted from increasing the price of the product by more than 10% over the preceding twelve months7 or having its ceiling price8 notified by the National Pharmaceutical Pricing Authority.
- Greater Oversight by the Health Ministry
The Health Ministry will also have more control over medical devices once they have been brought under the purview of the MDR. The Health Ministry can change or introduce new standards for regulating medical devices and even ban devices that are not proven safe and efficacious for patient use.9 The CDSCO will have the power to inspect premises where medical devices are manufactured and sold to ensure that such premises are as per standards prescribed by law.
3.Overview of the Notification
The Notifications were both released on the same day and would also come into effect on the same day i.e. April 01, 2020. The Notifications are largely based on the roadmap for medical devices discussed by the Drugs Technical Advisory Board – India’s apex body on technical matters related to drugs –in April 2018, and the Draft Notifications published thereafter.10
Once the New Definition Notification comes into force, the MDR will apply to all medical devices except for the devices in Annexure A (“New Devices”). Annexure A contains the list of medical devices that have been specifically notified thus far.
Interestingly, the New Definition Notification does not change the existing definition of medical devices under section 3(b)(iv) of the D&C Act.11 Instead, the New Definition Notification notifies a catch-all definition of medical devices, effectively bringing all medical devices under the regulation of the MDR as follows:
“All devices including an instrument, apparatus, appliance, implant, material or other article; whether used alone or in combination, including a software or an accessory, intended by its manufacturer to be used specially for human beings or animals which does not achieve the primary intended action in or on human body or animals by any pharmacological or immunological or metabolic means, but which may assist in its intended function by such means for one or more of the specific purposes of -
- diagnosis, prevention, monitoring, treatment or alleviation of any disease or disorder;
- diagnosis, monitoring, treatment, alleviation or assistance for, any injury or disability;
- investigation, replacement or modification or support of the anatomy or of a physiological process;
- supporting or sustaining life;
- disinfection of medical devices; and
- control of conception.”
Therefore, once the New Definition Notification comes into effect, manufacturers and importers of New Devices would be required to obtain manufacturing and import licenses under the MDR to engage in the manufacture and import of medical devices.
However, once the New Device has been registered on the Portal as per the Procedure described in the Registration Notification, such device will be exempt from the MDR (and consequently from obtaining the above-mentioned manufacturing and import licenses) for a period of 30 months from April 01, 2020 if the New Device is a Class A or Class B device, and for a period of 42 months if the New Device is a class C or Class D device (“Exemption Provision”).
Notably, the registration of New Devices on the Portal is on an optional basis for 18 months and mandatory thereafter. However, as the Exemption Provision is applicable only for New Devices that have been registered on the Portal, manufacturers and importers have a strong incentive to register their New Devices as soon as the Portal becomes operative so that they can avail of the Exemption Provision.
The details required to be uploaded by manufacturers and importers of the New Devices are specified in Annexure 2.
C. ANALYSIS
The Notification is the biggest leap that India has taken in terms of medical device regulation after the enactment of the MDR in 2017. However, there are some concerns with respect to both clarity and expected impact with respect to the Notifications, as outlined below.
- Tight Timelines for Ensuring Compliance
Registering the New Devices is a pre-condition to availing the Exemption Provision. Therefore, manufacturers and importers will be compelled to register the New Devices to avail the Exemption Provision or not import/manufacture the New Device until they obtain the requisite licenses. In the event the Portal is not operational on time or experiences technical difficulties, medical device companies may be forced to halt operations entirely after April 01, 2020 until they are able to register their medical device.
Alongside the registration process, manufacturers and importers will be required to apply for manufacturing and import licenses with the CDSCO, in order to comply with the larger ambit of the MDR once the Exemption Provision expires – a process that typically takes nine months to complete. However, given the large volume of applications the CDSCO is bound to receive, processing the applications may take even longer. This issue may be further compounded as the CDSCO is currently inadequately staffed to deal with such a large volume of applications.
As a result, manufacturers and importers who have not been granted the required licenses at the expiry of the Exemption Provision despite having registered their New Devices and applied for licenses well in time will be forced to halt operations.
While halting business operations is certainly a concern from a business perspective, the more significant ramification is from a patient safety perspective. Gaps in supply of medical devices could leave patients without the care they need and can even be life threatening in cases where patients need devices such as pacemakers.
Ideally, the Exemption Provision should have been de-linked from the requirement to register the device. This way, New Device manufacturers/importers would have had the full 30 or 42 months to register their New Device. Additionally, manufacturers and importers who have registered their New Device on the Portal and applied for a manufacturing/import license should have been permitted to carry on business activities on a provisional basis at the expiry of the Exemption Provision, until either a license is granted or the application is rejected. This may have helped stagger the workload on the CDSCO without requiring medical device companies to halt their business activities.
- Ambiguities on Registration Process
The details required to be uploaded by the manufacturer and importer (specified in Annexure 2) contemplate the manufacture or import of a medical device as a whole. However, a medical device could also be partly manufactured in India and partly imported. Additionally, spare parts of medical devices may also be imported separately for repairing the medical device. Clarification is still required from the CDSCO on how registration numbers will be generated for medical devices which cannot be registered as a whole.
The manufacturer or importer is also required to specify the class of the medical device sought to be manufactured or imported when entering the details of the medical device on the Portal. The CDSCO will therefore be required to classify a large number of medical devices before the registration requirement comes into effect which, given the impending timeline, appears to be a herculean task. The CDSCO should ideally clarify whether importers/manufacturers can self-classify based on similar classifications in other jurisdictions, in order to ease the transition.
Additionally, before the Exemption Provision expires, the CDSCO would also be required to ascertain which of the medical devices registered on the Portal may be considered to be investigational medical devices12/new in-vitro diagnostic devices.13 Broadly, this would require the CDSCO to look into whether each device registered on the Portal has a history of safe use in India, or whether the medical device needs to undergo clinical testing before it can be considered to be safe and efficacious for use in India. It is currently unclear whether existing devices in the market, although sold for a long time in India, would be considered to be investigational devices, given that safety and efficacy data was not required to be generated when the device was first launched. Due to this, it is possible that the first applicant of each category of medical device would be required to undergo clinical evaluations before the device can be re-introduced into the market. Effectively, this means that there could be an availability gap of numerous devices, until approvals are granted for the re-introduction of the device post the expiry of the Exemption Provision. Ideally, the CDSCO should clarify the parameters to determine when a New Device would be considered to be an investigational medical device.
To add to the ambiguity, as of this writing, the Portal i.e. the Online System for Medical Devices does not appear to provide for registration of medical devices. It is therefore unclear whether the registration process would start only once the Notifications come into effect, in which case, importers and manufacturers would be required to wait until the registration is issued post April 2020 (unless the entire process as envisaged under the Notifications runs smoothly and registration numbers are auto-generated immediately upon submission of the application), before commercial activities can continue. Hopefully, the Portal would be operational soon so that medical device manufacturers and importers can register their devices by the time the Notifications come into effect. Alternatively, the CDSCO should allow for a grace period after April 2020, wherein medical device manufacturers/importers who have made the application for registration can continue to manufacture/import, until such time a registration is granted.
- Status of Medical Devices Exempted under the Registration Notification
The medical devices that have already come under the purview of the MDR or will be coming under the purview of the MDR over the next year have been specifically excluded under the Registration Notification. As a result, these devices are not required to be registered on the Portal. Further, these devices would also be exempt from compliances under the MDR until the date the medical device is slated to come under regulation. For instance, MRI equipment which has been notified under the MDR with effect from April 01, 2021 would not be governed under the MDR by virtue the Notifications (and therefore no registration of MRI equipment would be required).
However, MRI Equipment would be governed by the MDR with effect from April 01, 2021. Therefore, manufactures and importers of MRI Equipment would be required to obtain the requisite licenses prior to April 01, 2021 to carry on commercial activity relating to MRI Equipment thereafter. Typically, the regulators take anywhere between 9 to 12 months to grant an import/manufacturing license. Therefore, the CDSCO and state licensing authorities should start accepting applications for the granting manufacture/import licenses in respect of the devices in Annexure A which are yet to come under regulation. A delay in obtaining/grant of license would force the manufactures/importers of these devices to cease business operations in respect of the device and would lead to a loss in business and revenue for the medical device manufacturers.
D. CONCLUSION
The Notifications are clearly a step in the right direction. Bringing medical devices under MDR will lead to improvements in the quality of medical devices and consequently an increase in patient safety. Wider medical device regulation will also lead to uniformity in medical device standards across all brands and manufacturers thereby reducing danger to patient safety and decreasing the need for constant medical supervision. Manufacturers will also have a definite set of standards to comply with as opposed to attempt to set their own methods of validating medical devices.
However, while the intention of the Notifications is laudable, clarifications are required from the CDSCO on how the implementation will take place. Effective implementation may lead to gains for patients while delayed or ineffective implementation could lead to huge losses for medical device companies. It also bears mentioning here that despite having received comments from various industry stakeholders on the Draft Notification, no industry comments have been incorporated in the Notifications.
Even aside from the Notifications, the medical device regulation space has been experiencing a lot of activity. Earlier in November 2019, the NITI Aayog – the Indian Government’s policy think tank – submitted a draft bill to the Indian Government proposing to create a separate regulator for medical devices. The bill, titled the Medical Devices (Safety, Effectiveness and Innovation) Bill, 2019 also aims to introduce a Unique Identification Number on all medical devices, increase penalties for non-compliance as well as tighten the regulation applicable to clinical investigations.14 Therefore, it is peculiar how there is simultaneously a push towards regulating medical devices under a separate legislation and regulator, and towards bringing all devices under the ambit of the MDR. Nonetheless, the effectiveness of the Notification is in its implementation and in a few months, the proof will be in the pudding.
– Shreya Shenolikar, Darren Punnen & Dr.Milind Antani
You can direct your queries or comments to the authors
1 https://cdsco.gov.in/opencms/opencms/system/modules/CDSCO.WEB/elements/download_file_division.jsp?
2 https://cdsco.gov.in/opencms/opencms/system/modules/CDSCO.WEB/elements/download_file_division.jsp?
3 Rule 4 MDR
4 https://www.ncbi.nlm.nih.gov/books/NBK222708/
5 Rule 7 MDR.
6 Fifth Schedule MDR.
7 Paragraph 20 DPCO.
8 Paragraph 4 DPCO.
9 S.10A D&C Act.
10 https://cdsco.gov.in/opencms/opencms/system/modules/CDSCO.WEB/elements/common_download.jsp?
11 S.3(b) “drug” includes –
(iv) such devices intended for internal or external use in the diagnosis, treatment, mitigation or prevention of
disease or disorder in human beings or animals, as may be specified from time to time by the Central Government by notification in the Official Gazette, after consultation with the Board.
12 Investigational Medical Device is a device which (i) does not have a predicate device i.e. the device is
first of its kind, or (ii) has been approved but is now sought to be launched for a new intended use or new population or new material or major design change.
13 “new in vitro diagnostic medical device” means any medical device used for in vitro diagnosis that has not
been approved for manufacture for sale or for import by the Central Licensing Authority and is being tested to establish its performance for relevant analyte or other parameter related thereto including details of technology and procedure required.